应用数学与交叉科学研究中心生物信息学团队于2024年3月第3次组会按期举行,小组全体成员和各位导师共同参加。在这次组会上,由一名研二员工和六名研三员工分别汇报自己的研究进展,然后老师与同学们对汇报内容进行学术探讨,并对存在的问题给出相应的指导和建议。
王梦园:本次汇报了毕业论文的主要内容。论文题目为:《基于单细胞转录组测序数据的基因转录爆发动力学推荐》,文章主要介绍了两个主要工作:1.研究总结并分析了热门的单细胞转录组测序数据缺失值插补方法,并给出推荐方案;2.通过数据驱动和模型驱动相结合的模式结合单细胞转录组测序数据与基因表达模型推断转录爆发动力学特征,并在模拟数据集和真实数据集上进行验证,最后分析了基因表达和转录爆发的内在关系。
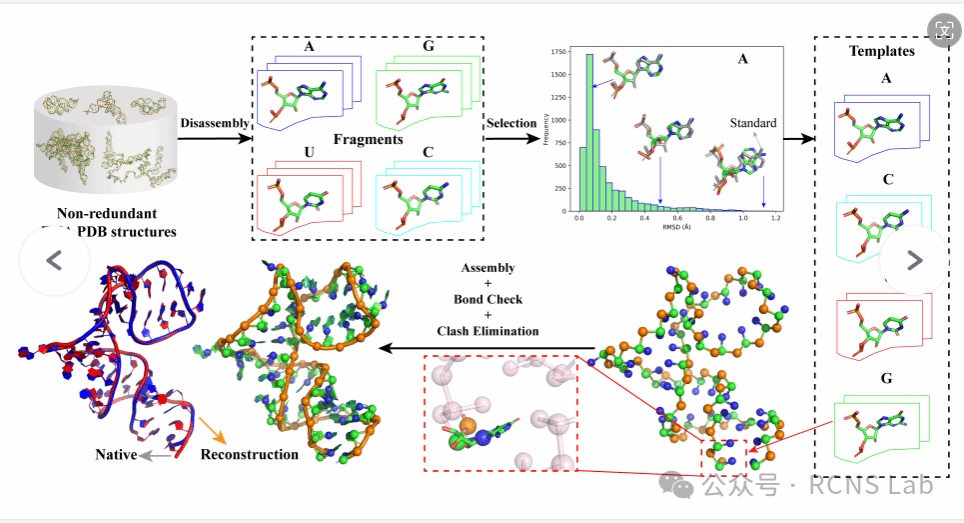
吴昊:本次汇报了毕业论文的研究内容。RNA分子具有重要的生物学功能,然而现有的实验方法费时费力,虽然基于物理原理的粗粒化模型可以较为快速的预测出RNA结构的粗粒化结构,但粗粒化模型缺乏原子细节,这极大的限制了它的应用性。现有的几种全原子还原方法又各自存在一定的局限性,基于现状我们提出了一种新的基于多核苷酸片段组装的直接方法,以快速、准确地实现粗粒化结构的全原子还原。
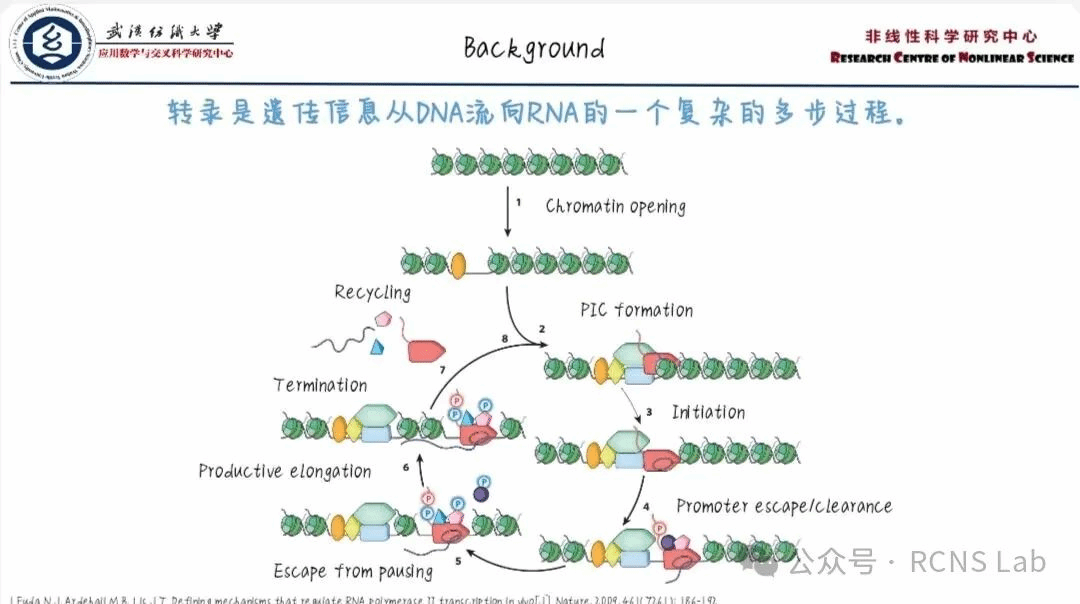
韩长锋:本次汇报了毕业论文的研究内容,如下:细胞命运决定是胚胎发育和多细胞生物形成的关键过程,研究细胞命运决定的机制对理解细胞内部调控至关重要。细胞增殖,分化和重编程等细胞过程由复杂的基因调控程序控制,但细胞通过整合各种信号并执行基因调控指令做出自己的命运决定。本研究旨在深入探究细胞命运决策的内在机制,特别是影响基因表达量变化的关键因素。与传统的细胞命运决策研究路线不同,本文拟采用模型与数据相结合的方法,综合深度学习和化学主方程等数学计算机方法,对细胞命运决策过程进行了系统的分析。
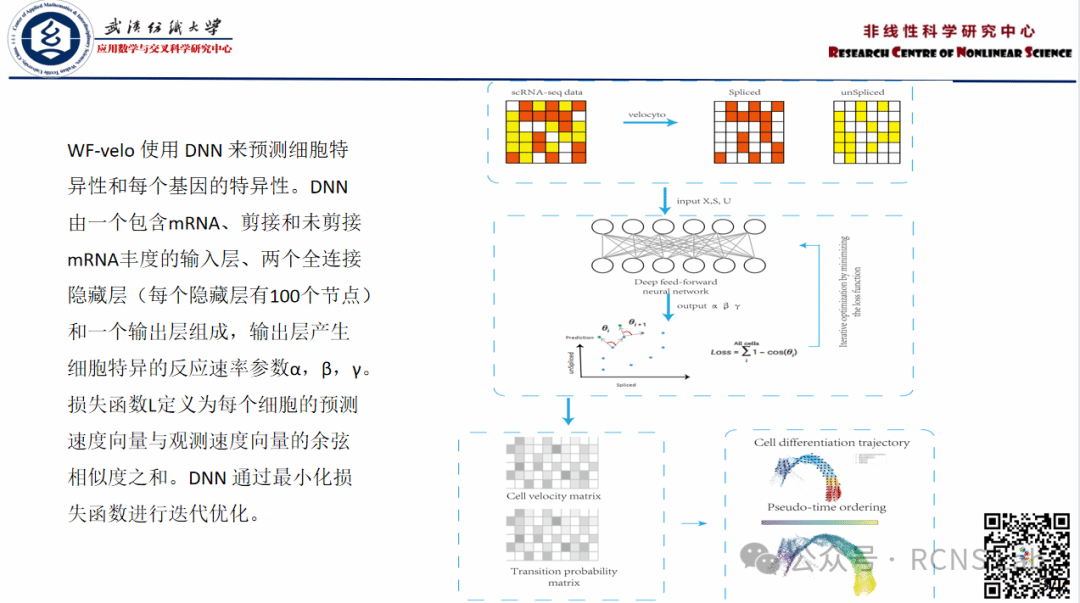
李成:本次汇报了毕业论文的研究内容。为了能够解决缓解估计定量未剪接mRNA和剪接mRNA丰度对估计RNA速度的影响,并且不需要复杂的额外的特征信息来增强RNA速度的估计,我们改进了RNA速度模型,设计一个新的RNA速度模型WF-velo,具有简单并能提高单细胞测序数据的利用率,用完整的细胞基因表达特征信息来估计RNA速度,并推断细胞分化轨迹。
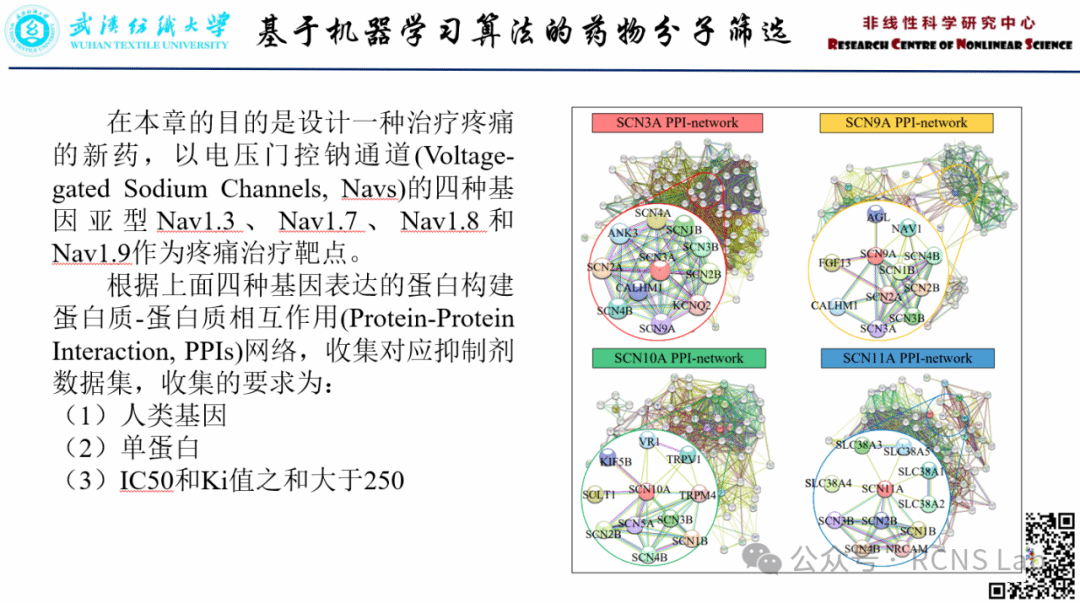
窦博正:本次汇报了毕业论文的研究内容。药物分子数据,特别是疼痛及药物成瘾相关的数据涉及多种药物分子属性及研究内容,这些数据对于药物研发、药物活性预测和药物分子虚拟筛选等研究至关重要。随着人工智能技术的快速发展,机器学习算法在药物分子数据分析方面具有广泛的应用。此外,以代数拓扑理论为核心的拓扑数据分析方法在药物设计领域取得巨大成功,提高了药物分子虚拟筛选的准确度和效率。因此,我们结合机器学习、深度学习以及代数拓扑,通过收集和分析药物成瘾数据和疼痛相关分子数据,筛选治疗药物成瘾和疼痛的先导化合物分子,并详细介绍了用于处理小样本药物数据集的机器学习方法。
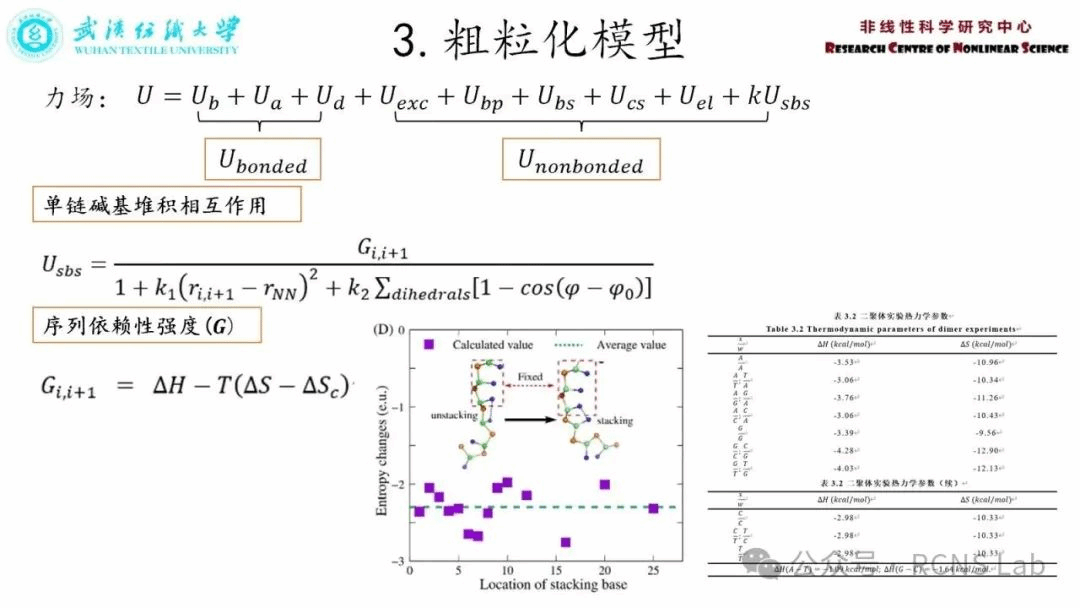
袁洁:本次汇报了毕业论文的研究内容。针对以前开发的一个DNA粗粒化模型有进一步的发展,增加了序列效应依赖的单链内相邻碱基之间的堆积相互作用,以预测单链DNA结构及其柔性中的序列效应。为了验证该模型,针对不同长度(8-100nt)的单链Poly(dA)和Poly(dT),利用该模型预测了其在不同离子条件下的构象变化,并计算回转半径随序列长度及离子浓度的变化关系。还进一步利用该模型研究了序列及长度、离子浓度等对单链DNA的持久长度的影响。
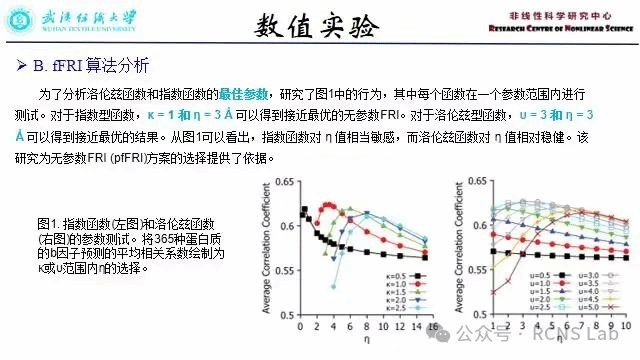
柯璐:本次汇报了一篇文献《Fast and anisotropic flexibility-rigidity index for protein flexibility and fluctuation analysis》,使用适当的数据集引入快速 FRI (fFRI) 算法,与原始 FRI 算法的 O(N2) 和 GNM 的 O(N3) 相比,所提出的 fFRI 的计算复杂度为 O(N),其中 N 是原子数。另外,引入各向异性 FRI (aFRI) 算法来进行生物分子的运动分析。与完全全局且黑塞矩阵中有 3N × 3N 元素的 ANM 不同,所提出的 aFRI 算法具有自适应 黑塞矩阵,其范围从完全全局到完全局部。尽管存在局部化,但三组特征向量中仍存在集体运动。此外,进一步分析 FRI 方法在蛋白质 B 因子预测方面的性能。为此,检查了与四个相关函数相关的 FRI 算法的准确性,并对 FRI 和 fFRI 与其他前沿方法(即 NMA 和 GNM)进行了比较研究。
— 员工汇报照片展示 —




