应用数学与交叉科学研究中心生物信息学团队于2023年11月21日举行第二十八期研究生论坛,小组全体成员和各位导师共同参加。在这次组会上,由两名研一员工和两名研二员工分别汇报自己的研究进展,然后老师与同学们对汇报内容进行学术探讨,并对存在的问题给出相应的指导和建议。
郭成:本次汇报一篇文献《Selection of sequence motifs and generative Hopfield-Potts models for protein families》进化相关蛋白质家族的统计模型最近引起了人们的兴趣,特别是,通过直接耦合分析(DCA)推断的成对Potts模型能够提取折叠蛋白质的三维结构和蛋白质中氨基酸取代的信息。在此,本文提出了一种基于选择集体序列基序(motifs)的参数约简方法。它自然地导致了根据霍普菲尔德-波茨(Hopfield-Potts)模型建立的统计序列模型的形成。这些模型可以精确地推断使用映射到限制玻尔兹曼机器(RBM)和持久的对比散度(PCD)。本文表明,当应用于蛋白质数据时,即使是20-40个模式也足以获得统计上接近生成的模型。Hopfield模式形成可解释的序列基序,并可用于将氨基酸序列聚类为功能亚家族。

张秋玉:本次汇报了一篇文献《A mechanistic model for the negative binomial distribution of single-cell mRNA counts》。在分析单细胞RNA测序实验的结果时,通常需要假设观察到的测序计数的概率分布。哪种是最合适的离散分布是一个悬而未决的问题。为了解决可解释性的问题,研究了常用的离散概率分布下的机制转录和降解模型:从已知的转录降解模型推断稳态概率分布,如Poisson分布或Poisson-beta分布。把这个过程颠倒,我们展示了如何从给定的概率分布推断出相应的生物模型,这里是负二项分布。到目前为止,支持这一分布假设的机制模型尚不清楚。文章的结果表明,负二项分布是以稳态分布的形式出现的,它来自于一个爆发产生mRNA分子的动力学模型。我们的经验表明,它在计算复杂性和生物简单性之间提供了一种方便的权衡。
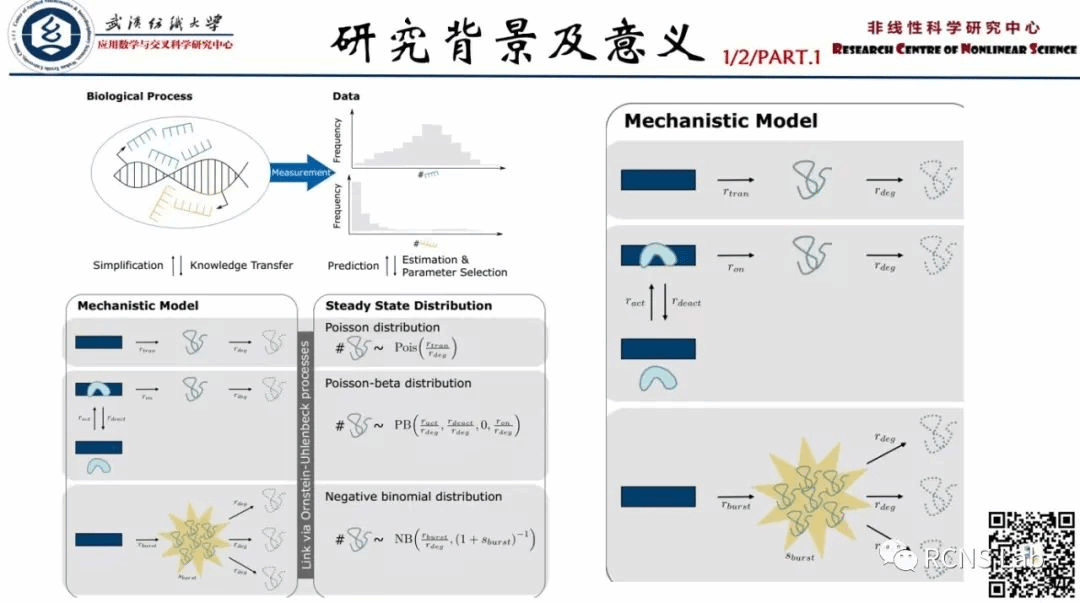
万智康:本次汇报了一篇文献《RNA secondary structure prediction using an ensemble of two-dimensional deep neural networks and transfer learning》,人类基因组的大部分转录成结构和功能未知的非编码RNA。获得非编码RNA的功能线索需要精确的碱基修复或二级结构预测。然而,目前基于折叠的算法在这种预测方面的表现已经停滞了十多年。在这里,我们建议使用深度上下文学习来预测碱基对,包括那些由三级相互作用稳定的非规范和非嵌套(伪结)碱基对。由于只有<250个非冗余的高分辨率RNA结构可用于模型训练,我们利用迁移学习从一个模型中进行初始训练,该模型最初是用最近的高质量bpRNA数据集训练的,该数据集包含了通过比较分析获得的大于10万个非冗余RNA。所得到的方法在预测所有碱基对,特别是非规范和非嵌套碱基对方面取得了统计学上显著的进步。该方法(SPOT-RNA)具有免费的服务器和独立软件,可用于改进RNA结构建模、序列比对和功能注释。
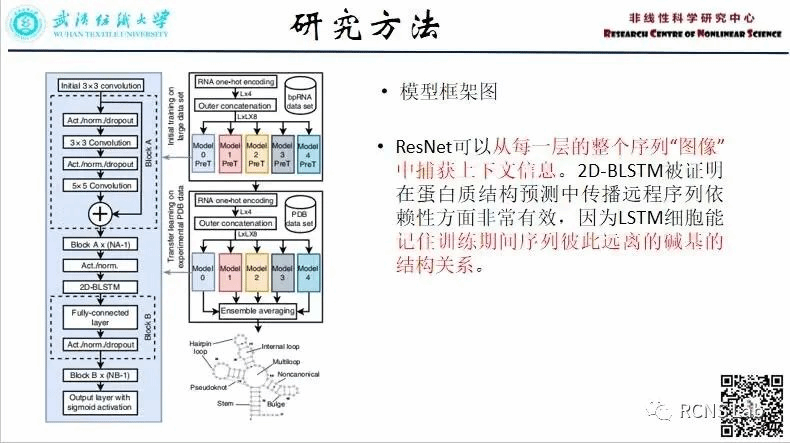
王永康:本次组会汇报了一篇文献《Molecular generative model based on conditional variational autoencoder for de novo molecular design》。其中使用ZINC开源数据库中的50万个分子数据,在SMILES 代码末尾填充了一个“E”以指示字符串的结尾,同时将本实验设置的五个目标属性来构成条件向量,并通过编码器中RNN生成和组合潜在空间向量和条件向量,然后在解码器中使用softmax层来优化CVAE函数。解码器单元的每个输出向量表示 SMILES 代码字符和“E”的概率分布。最后,输出向量被转换为 SMILES 代码。本文提出了一种基于条件变分自动编码器的新型分子设计策略。作为深度学习生成模型之一,该方法的优势在于能够通过将多个目标属性应用于条件向量上来同时控制它们。此外,我们可以在不改变其他属性的情况下,有选择地调控某一目标特性的数值,并且拓展了对特定属性的训练集范围。
— 员工汇报照片展示 —



